Flowchart
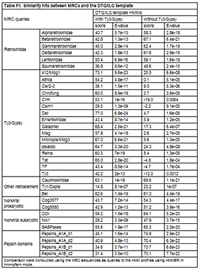
The clan AA Reference Database (CAARD)The clan AA Reference Database (CAARD) is an attempt at characterizing the clan AA protein domain in all its possible phylogenetic signals by ancestral maximum likelihood reconstruction (AMLRs), sequence logos and hidden Markov models (HMMs). With this aim, we performed a comprehensive study of 323 non-redundant clan AA peptidases (CAPs) dividing them into 38 protein families according to prior estimates of their taxonomy and relationships (for more details see Table 1 in Llorens et. al.). In this section we describe the bioinformatic flowchart designed to normalize and characterize the different signals. The flowchart can be divided in 4 steps, a description of each step follows. Multiple alignments and consensus identificationBecause the different families present in the current database were originally assumed from prior investigations, we compared all sequences to each other via the BLAST search at GyDB using the CORES Database in BLASTp mode. With very few exceptions, the BLAST analysis confirmed that each family follows a particular phylogenetic signal, or in other words, that the sequences belonging to each family are usually more similar to one another than to other sequences (data not shown). From that point on, we created a set of 34 alignments, one for each family that includes 2 or more CAPs to characterize the protein domain architecture of each family (we dismissed the CAPs representing one-sequence families from the 38 families summarized in Table 1 of Llorens et. al.). Additionally, we performed a non-redundant single multiple alignment with all CAPs to evaluate the phylogeny and major consensus. Here, we used prior structure-based alignments (Pearl and Taylor 1987; Weber 1989) and Andreeva's model (Andreeva 1991) as the criterion to manually align the different families. The alignment was refined using a conventional phylogeny as guide tree. While performing this alignment, we identified 6 amino acid patterns with structural correspondence with Andreeva's model. We will refer to these patterns as the DTG/ILG template because of the DT/SG and ILG amino acid motifs (Pearl and Blundell 1984; Pearl and Taylor 1987) usually found in all CAPs are prominent. Alignments are freely available in the GyDB collection deposited in Biotechvana Bioinformatics (for more details see Llorens et al.). Information content enhancementClan AA is an extremely difficult case study of fast evolving protein. Software algorithms fail to align exhaustively the different clan AA families and the degree of sequence preservation varies depending on the family. This means that while many families show high degree of preservation, others have not sufficient information content by themselves. To overcome this limitation, we used each family alignment as an input to FastML (Pupko et al 2000) to reconstruct by phylogenetic means a set of AMLR sequences by 2 alternative methods, Joint and Marginal (Aldrich 1997; Koshi and Goldstein 1996; Pupko, Pe'er, Shamir, and Graur 2000; Yang et al 1995). The AMLR analysis provides several files with information about different aspects of the AMLR analysis (this includes 2 Jrof and Mrof alignments between CAPs and AMLR sequences). Here, we do not try to reconstruct the single ancestor of each family (statistically, this probability approaches 0), but a variety of ancestral states to enhance the information content of each family alignment. With this, the object of the AMLR analysis is to increase the most prominent sequence patterns specifically preserved by each set of monophyletic CAPs (the main principle we tested in previous steps is, sequences belonging to a monophyletic family are more similar to each other than any other sequence). HMMs and sequence logosWe selected the AMLR alignments, removed the non-informative traits from all of them, and used those obtained with the Jrof method to create a collection of HMM profiles and sequence logos, with HMMER (HMMER 2008) and CheckAlign, respectively. We used the processed AMLR alignments instead of conventional alignments to minimize conflicting signals among families in the HMMs, and build the sequence logos taking advantage of the information content enhancement. Finally, we constructed a set of MRC sequences derived from the HMMs using HMMER. HMMs and the MRCs were tested using the COREs database via the BLAST and HMM searches at GyDB, in BLASTp and hmmsearch, respectively. Major consensusWe tested different tools to characterize the DTG/ILG template as a sequence logo or as an HMM profile using the non-redundant alignment as input. Because of the variability and multiple gaps introduced in the alignment, all tools failed to reconstruct informative material. In an attempt to increase the information content of the non-redundant alignment, we conducted an additional AMLR analysis using this alignment as an input to FastML. This strategy also failed to obtain an AMLR alignment with sufficient information content for creating a sequence logo or an HMM. Then, we tried an alternative strategy, anchoring the MRCs derived from HMMs in a single master alignment manually based on the 6 DTG/ILG template's patterns. To cover all possible sequence states in this alignment we also aligned the LTRCAPs representing one-sequence families dismissed from the set of alignments. We performed an additional AMLR analysis using this alignment as an input to FastML and removed the non-informative traits from the resultant AMLR alignments (as we did with the other AMLR alignments used to create HMMs and sequences logos). In the Figure P1 within this section, we show the resolved Jrof alignment from this analysis, after the processing of non-informative traits. Again, we would stress that performing AMLR we did not try to resolve any kind of relationship between input and AMLR sequences. The goal was to enhance the most prominent sequence patterns of the master alignment to build an informative computational material. Taking into account the large number of Ty3/Gypsy MRCs used in the analysis, we conducted a second AMLR analysis excluding all these sequences from the master alignment. Figure P2 shows the Jrof alignments resolved from this alternative AMLR analysis, after the processing of non-informative traits. Both AMLRs had sufficient information content to create a sequence logo and an HMM profile. We tested the 2 HMMs with the different MRCs involved in their reconstruction and with all CAPs originally classified using the HMM sever available at GyDB. Both HMMs showed strong similarity to the different MRCs (Table P1) and displayed a wide range of similarity detection when comparing them to CAPs. However, the HMM implemented by Ty3/Gypsy sequences proved more capable than the alternative HMM, as it covers more states than the alternative model. Cite this bioinformatic flowchart and DatabaseLlorens,C. Futami, R., Renaud, G. and A. Moya. Bioinformatic Flowchart and database to investigate the origins and diversity of clan AA peptidases. (manuscript accepted, Biology Direct) References
|
|